DCARSacrossNetwork
DCARSacrossNetwork.Rdperforms DCARS method across all edges listed in the network for the (already ranked) genes x samples matrix dat
DCARSacrossNetwork(dat, edgelist, edgeNames = rownames(edgelist), ...)
Arguments
| dat | a genes x samples gene expression rank matrix, should be already converted to ranks with first column lowest survival and last column highest survival |
|---|---|
| edgelist | is a 2 column character matrix with the genes to test for DCARS. if edgelist has more than 2 columns then the first two are taken |
| edgeNames | is the name to assign to each edge, defaults to rownames(edgelist). if edgelist has no rownames then defaults to two gene names pasted with "_" in alphabetical order |
| ... | additional parameters passed to DCARS() |
Value
value of this function depends on arguments passed into the DCARS() function (e.g. if extractTestStatisticOnly is set as TRUE)
Examples
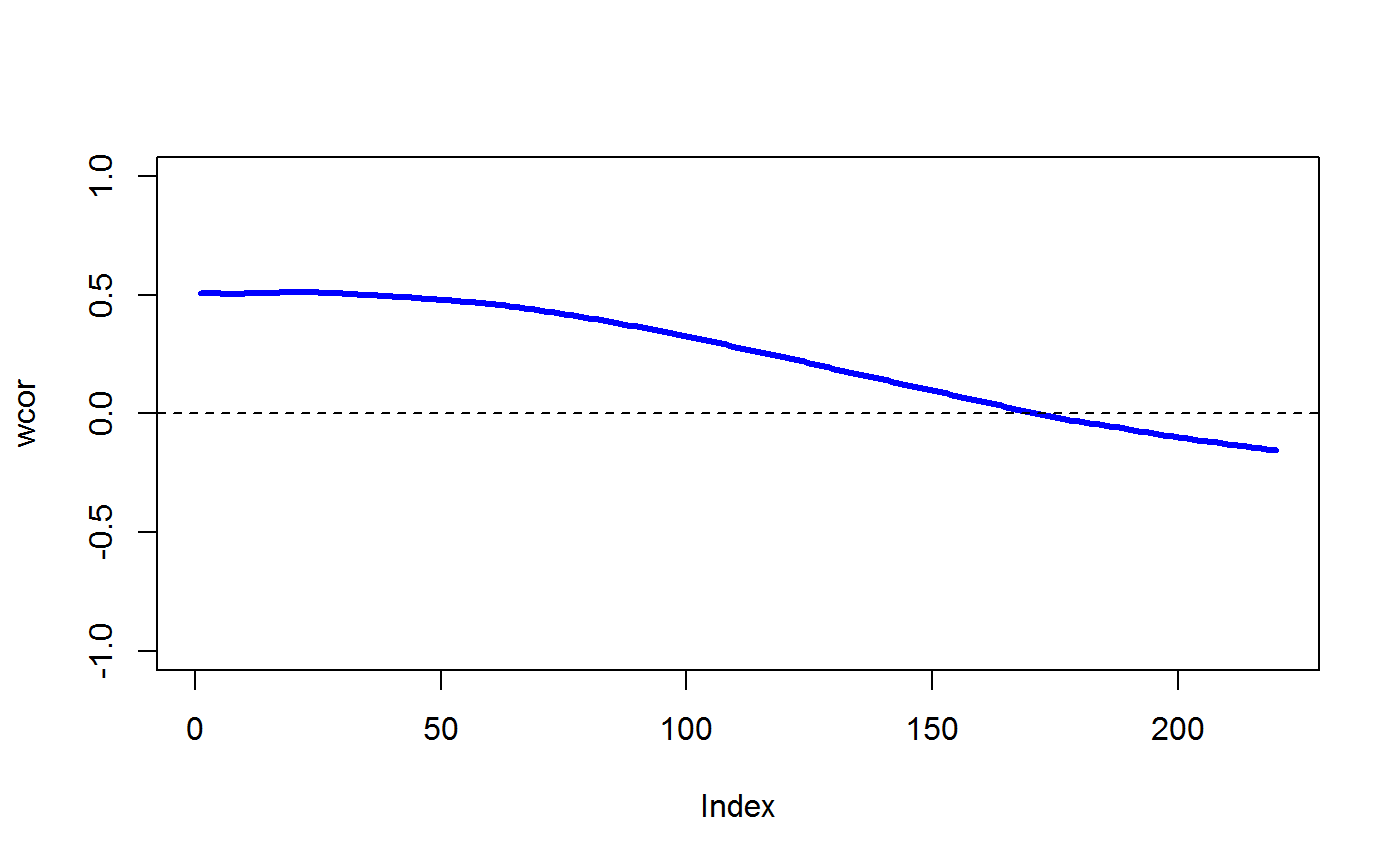
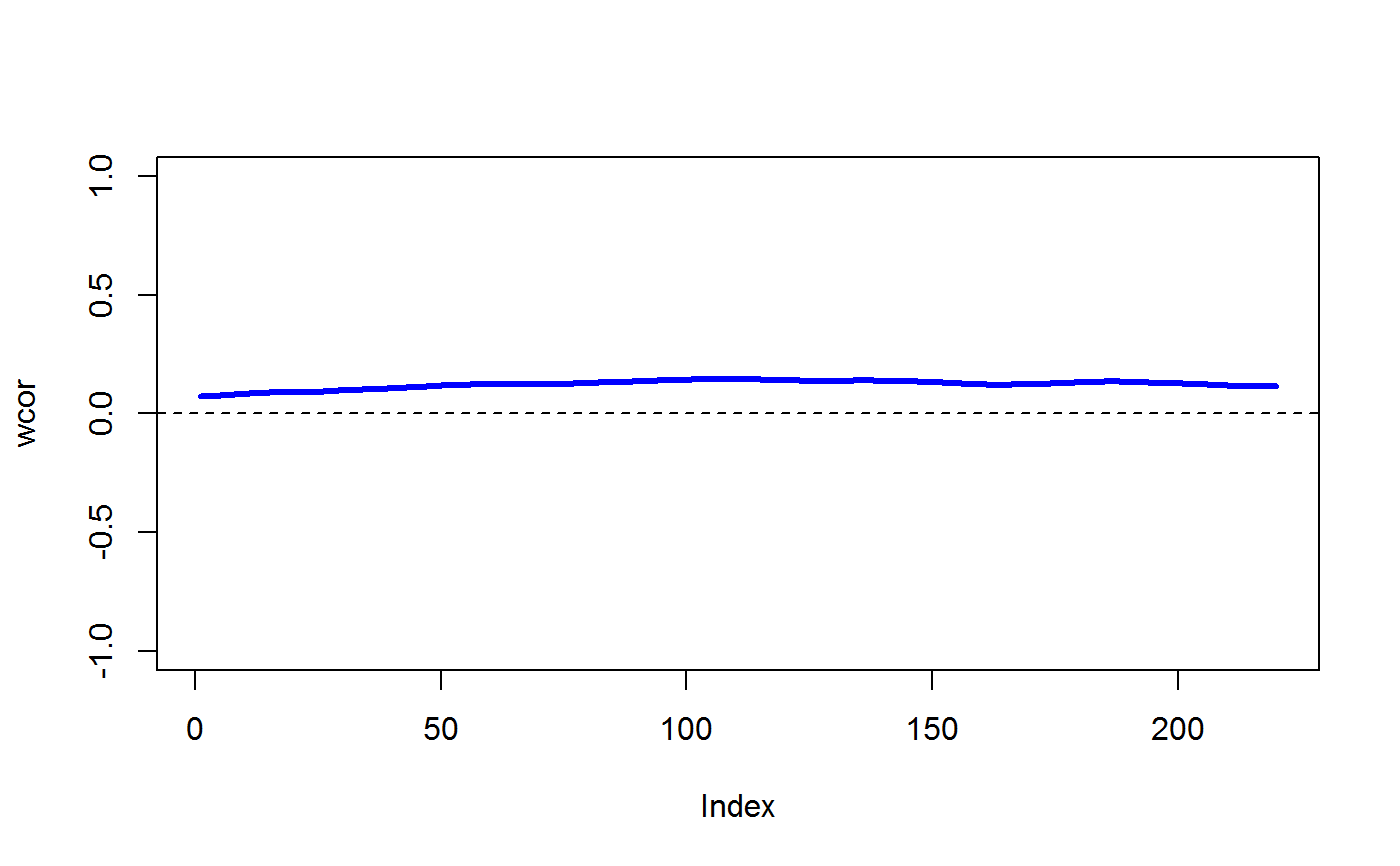
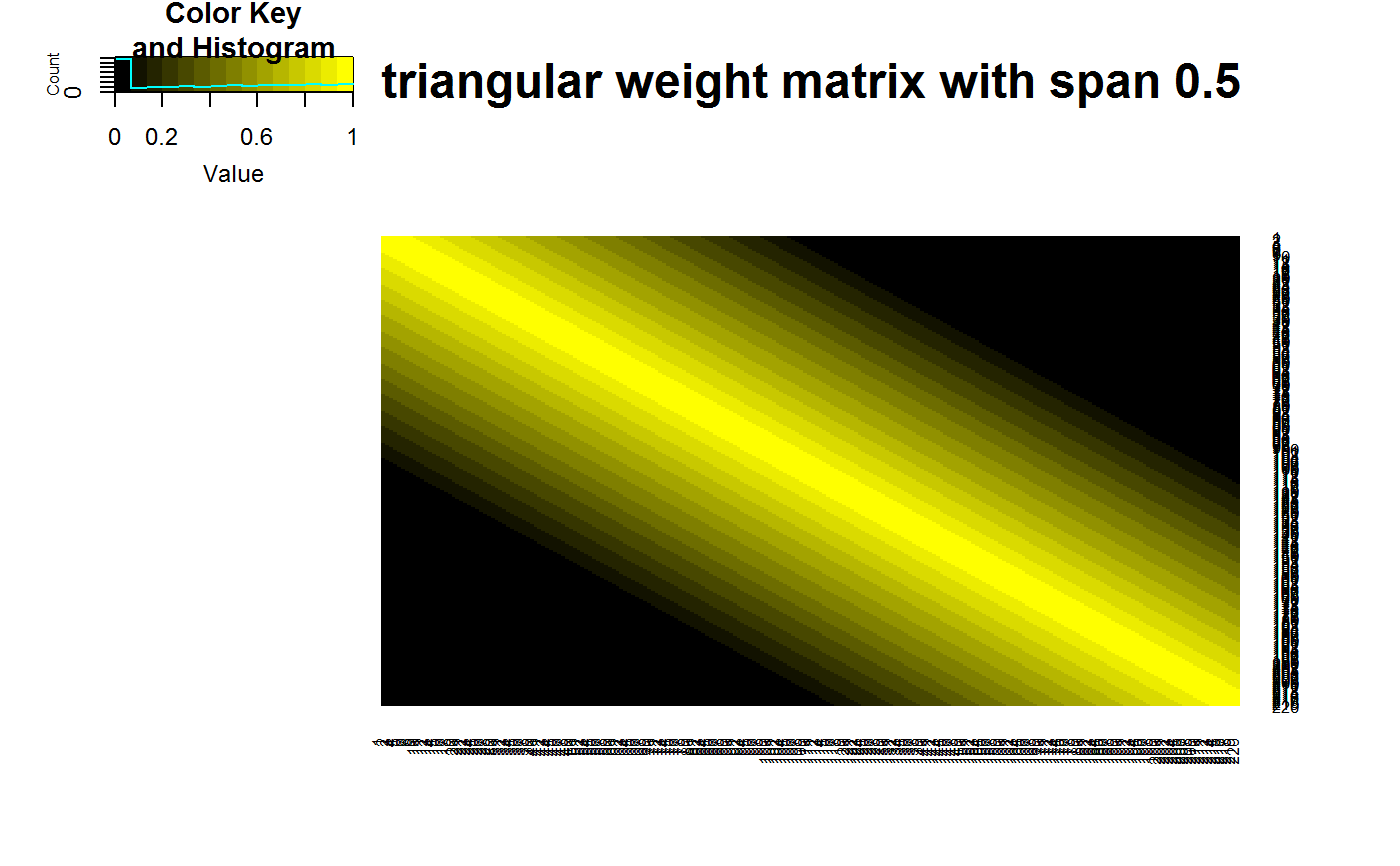
data(STRING) data(SKCM) SKCM_rank = t(apply(SKCM,1,rank)) # highly significantly DCARS gene pair: SKP1 and SKP2 # calculates p-value based on permutation DCARS(SKCM_rank,"SKP1","SKP2",plot=TRUE)#>#>#>#> [1] 0#>#>#> [1] 0.227379# not significantly DCARS gene pair: EIF3C and EIF5B # calculates p-value based on permutation DCARS(SKCM_rank,"EIF3C","EIF5B",plot=TRUE)#>#>#>#> [1] 0.96#>#>#> [1] 0.01790925# build weight matrix W = weightMatrix(ncol(SKCM_rank), type = "triangular", span = 0.5, plot = TRUE)# extract DCARS test statistics SKCM_stats = DCARSacrossNetwork(SKCM_rank,edgelist = STRING, W = W, extractTestStatisticOnly = TRUE, verbose = FALSE) sort(SKCM_stats,decreasing=TRUE)[1:10]#> GMPS_IMPDH1 SKP1_SKP2 COPB2_COPE ANAPC10_ANAPC5 TBL3_UTP18 #> 0.2518867 0.2273790 0.2075771 0.1800155 0.1672255 #> PSMD12_PSMD3 POLR3B_POLR3D RAD18_UBE2B SNRPB_SNRPG RPL26L1_RPL8 #> 0.1659466 0.1629938 0.1621321 0.1555048 0.1531930